
|
Author(s)
Gregory S Schultz
Glenn Ladwig
Annette Wysocki
|
Contents
|
|
Published:
August 2005
Last updated: August 2005 Revision: 1.0 |
Keywords: extracellular matrix (ECM); acute wounds; chronic wounds; proteoglycans; glycosaminoglycans; metalloproteinases; chemokines; wound bed preparation; wound dressings.
The extracellular matrix (ECM) is the largest component of normal skin and gives the skin its unique properties of elasticity, tensile strength and compressibility.
In acute wounds the provisional wound matrix, containing fibrin and fibronectin, provides a scaffolding to direct cells into the injury, as well as stimulating them to proliferate, differentiate and synthesise new ECM.
Chronic wounds contain increased levels of inflammatory cells, giving rise to elevated levels of proteases that appear to degrade the ECM components, growth factors and receptors that are essential for healing.
Understanding of the importance of re-establishing a functional ECM in chronic wounds has led to technical advances and the development of products that reduce excessive protease levels or contribute functional ECM proteins, thereby facilitating the healing process.
The extracellular matrix (ECM) is the largest component of the dermal skin layer and the synthesis of ECM is a key feature of wound healing, especially when there has been a significant loss of tissue that precludes closure by primary intention. This article discusses the proteins contained in the ECM of normal skin that are important in the healing of acute and chronic wounds. In addition, the theory that the high levels of proteases found in chronic wounds impair healing by degrading essential components of the ECM is discussed using data from cell culture experiments and immunohistochemical analyses of wound biopsies.
As a consequence of this theory new dressings have been developed that are designed to reduce protease levels in wound fluids by providing a competitive substrate (collagen) for the proteases and thereby reducing proteolytic destruction of essential ECM components (fibronectin) and platelet-derived growth factors (PDGFs). Other new approaches include the topical treatment of chronic wounds with agents that reduce the synthesis of matrix metalloproteinases (MMPs), such as a mixture of metal cations, and treatment with unique proteins (amelogenin) to replace the corrupted ECM.
Clinical and laboratory data now clearly show that components of the ECM play an important role in normal wound healing and that the destruction of ECM components impairs healing. This has lead to the development of new therapies that aim to reduce the destruction of ECM or re-establish undamaged ECM.
The largest component of normal skin is the ECM, a gel-like matrix produced by the cells that it surrounds (Figure 1). The ECM is composed of a variety of polysaccharides, water and collagen proteins which give the skin remarkable properties [1], [2]. On a weight basis, the tensile (breaking) strength of normal skin approaches that of steel, yet skin also has substantial elasticity and compressibility. These properties are due to the combination of two main classes of ECM molecules, which are secreted by fibroblasts and epidermal cells. They are:
Fibrous structural proteins, including collagens, elastin and laminin, which give the ECM strength and resilience
Proteoglycans, such as dermatan sulfate and hyaluronan, typically consist of multiple glycosaminoglycan chains (formed from repeating disaccharide units) that branch from a linear protein core. Extracellular proteoglycans are large, highly hydrated molecules that help cushion cells in the ECM.
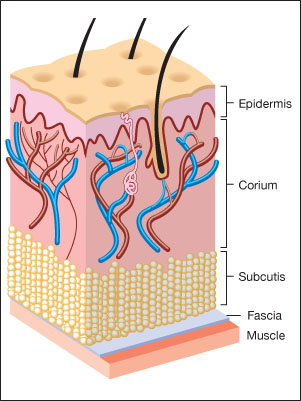
In addition to serving as a scaffold or structural support for cells, the ECM regulates cellular functions via cell adhesion, lubricates cells and provides a transport system for nutrients and waste products.
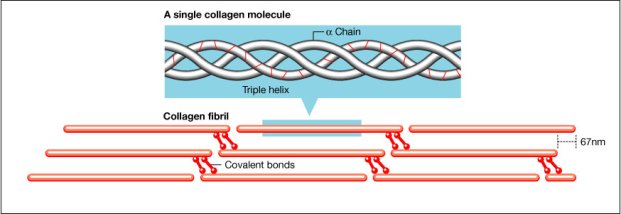
The largest class of fibrous ECM molecules is the collagen family, which includes at least 16 different types of collagen. Collagen in the dermal matrix is composed primarily of type I (80–85%) and type III (8–11%) collagens, both of which are fibrillar or rod-shaped collagens. The tensile strength of skin is due predominately to these fibrillar collagen molecules, which self-assemble into microfibrils in a head-to-tail and staggered side-to-side lateral arrangement. Collagen molecules become cross-linked to adjacent collagen molecules, creating additional strength and stability in collagen fibres(Figure 2).
Type IV collagen molecules associate with other specialised ECM molecules, including laminin and proteoglycans, to form a sheet-like structure known as the basement membrane (basal lamina), to which epidermal cells attach. This separates the epidermis from the dermis and surrounds blood vessels. Laminin is a large protein made up of three polypeptide chains that form a cross-shaped molecule. The four ends of the cross contain specialised regions that bind to type IV collagen molecules, heparan sulphate proteoglycan and specific proteins in the plasma membrane of cells called integrin receptors. This enables laminin to act as a multi-adhesive matrix protein by forming bridges between cells and the basement membrane.
Many tissues, such as skin, lung and blood vessels, need to be both strong and elastic to function properly. A network of elastic fibres in the ECM of these tissues gives them the required resilience to recoil after stretching. The main component of elastic fibres is the elastin molecule, which creates cross-links to adjacent elastin molecules. These molecules form a core of elastic fibres and are covered by fibrillin, a large glycoprotein that binds to elastin and is essential for the integrity of elastic fibres.
This is another important glycoprotein that is present in both plasma and tissue. Fibronectins have multiple functions, which allow them to interact with many extracellular substances, such as collagen, fibrin and heparin, and with specific membrane receptors on responsive cells.
Fibronectin has many different types of binding sites for collagen and fibrin molecules and heparan sulphate proteoglycan, allowing it to link together different types of ECM molecules. It also contains an important cell-binding domain made up of the three amino acids, Arg-Gly-Asp (RGD). Specific integrin receptors in the plasma membrane of cells recognise this RGD sequence of amino acids in fibronectin molecules. The binding of fibronectin molecules to integrin receptors on cells leads to the stimulation of signalling pathways that promote cell attachment, migration and differentiation. These characteristics enable fibronectin to play an important role in cell adhesion and to communicate signals between cells and components of the ECM.
Glycosaminoglycans (GAGs) are composed of polysaccharide chains made up of repeating disaccharide units and are strongly hydrophilic. GAGs are highly negatively charged and therefore attract osmotically active Na+, causing large amounts of water to be drawn into their structure. This results in GAGs occupying a huge volume relative to their mass and forming gels at very low concentrations. The hydrophilic nature of GAGs causes a swelling pressure, or turgor, which enables the ECM to withstand compression forces. The cartilage matrix lining the knee joint, for example, can support pressures of hundreds of atmospheres because of its high GAG content.
Hyaluronic acid (HA), a chief component of the ECM, is a large GAG that attracts water and is found in increased amounts in damaged or growing tissues. HA stimulates cytokine production by macrophages, thereby promoting angiogenesis.
Proteoglycans retain water and form a gel-like substance through which ions, hormones and nutrients can move freely. Given the great abundance and structural diversity of proteoglycan molecules, it would be surprising if their function was limited to providing lubrication around and between cells.
Perlecan is a proteoglycan present in the basement membrane, where it forms a gel of varying pore size and charge density. The epidermal cells of the skin derive their metabolic nutrients from the dermis via the diffusion of molecules through the basement membrane, so proteoglycans may play a key role in selectively filtering molecules that pass through the basement membrane beneath the epidermal cells.
Proteoglycans also appear to have an important role in regulating signalling between cells. For example, the heparin sulphate chains of proteoglycans bind to several different growth factors, including fibroblast growth factors (FGFs), helping them to bind to their specific cell-surface receptors[3], [4].
Proteoglycans can also bind chemokine molecules on the surface of endothelial cells, prolonging the inflammatory response. Although growth factors typically bind to the GAG chains of proteoglycans, members of the transforming growth factor beta (TGF-β) family bind to the core protein of the decorin proteoglycan. TGF-β directly increases scar formation by increasing the expression of collagen, fibronectin and lysyl oxidase while reducing the expression of MMPs, which break down the ECM. TGF-β is also chemotactic for macrophages and neutrophils, which prolongs the inflammatory response and contributes to scarring. When TGF-β is bound by the core protein of decorin it is not able to interact with its normal receptor protein on target cells, which inhibits the activity of TGF-β. Proteoglycans such as syndecan protect elastase from the inhibition by alpha-1 proteinase inhibitor, suggesting that they modify the proteolytic environment of wounds[5].
Acute wounds normally heal in an orderly and efficient manner by progressing through four distinct but overlapping phases: haemostasis, inflammation, proliferation and remodelling (Figure 3) [6],[7],[8]. Throughout these phases, components of the ECM play an important role in regulating and integrating many key processes of healing.
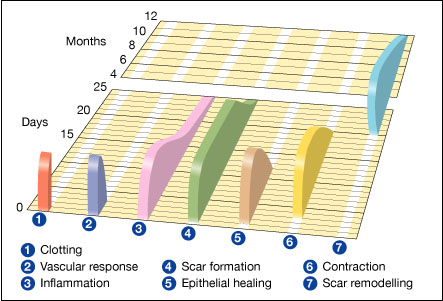
The clot that forms at the site of an injury is necessary to stop bleeding. However, it plays other important roles in wound healing by depositing at the wound interface a host of plasma and cell-secreted constituents. The epidermal cells subsequently dissect their way under the clot and over the granulation tissue, which is comprised of a dense population of macrophages, fibroblasts and newly formed blood vessels embedded in a loose matrix of fibrin, fibronectin, collagen and other ECM proteins. Stimulation of the clotting cascade results in the proteolytic cleavage of fibrinogen by the enzyme thrombin, forming an insoluble fibrin clot that holds damaged tissues together and provides the provisional matrix. In addition, the clot contains fibronectin molecules that are present in plasma and bind to fibrin through fibrin-specific binding sites.
About three days after injury fibroblasts begin to express new integrin receptors. On the fourth day, fibroblasts migrate into the provisional wound matrix as part of angiogenesis and in response to chemotactic growth factors released by platelets from the periwound area and subcutaneous tissue[9]. Using an in vitro model, fibronectin was found to be necessary for fibroblasts to express new integrin receptors and to migrate effectively over a collagen-fibrin matrix[10]. The integrin receptors then generate new intracellular signals that stop the fibroblasts migrating. Growth factors and proteins contained within the provisional wound matrix help to stimulate fibroblasts to begin proliferating and synthesising new collagen and other ECM components.
In this way the provisional wound matrix functions as much more than an inert scaffold in which scar tissue is deposited. It acts as a reservoir to help trap growth factors and actively signals fibroblasts, epidermal cells and vascular endothelial cells, via their integrin receptors, to transform into activated wound cells that will repair the injury.
Neutrophils: These are the first inflammatory cells to respond to the soluble mediators released by platelets and the coagulation cascade. They begin their journey from the vasculature into the injured tissue by first adhering to the vascular endothelial cell walls via their surface integrin receptors. They then release elastase and collagenase, which facilitate their migration through the basement membrane that surrounds the endothelial cells and into the ECM at the wound site. Their primary role is to mount the first line of defence against infection by phagocytosing and killing bacteria, and by breaking down foreign materials and devitalised tissue. Neutrophils also produce and release inflammatory mediators such as tumor necrosis factor alpha (TNF-α) and interleukin-1 (IL-1), which further recruit and activate fibroblasts and epithelial cells. Neutrophils produce and contain high levels of destructive proteases and oxygen free radicals, which they use to digest phagocytosed materials. On their inevitable death, neutrophils release these substances into the local wound area. This can cause extensive tissue damage and prolong the inflammatory phase. The persistent presence of high levels of bacteria in a wound may contribute to chronicity through continued recruitment of neutrophils and their release of proteases, cytokines and intracellular contents.
Monocytes/macrophages: Neutrophils are usually depleted in the wound after two to three days and are replaced by tissue macrophages. Macrophages begin as circulating monocytes that are attracted to the wound site, a process that begins about 24 hours after injury, by both soluble mediators and degraded components of the ECM, such as fragments of collagen and fibronectin. Monocytes bind to the ECM via integrin receptors and immediately differentiate into tissue macrophages. Serum factors and fibronectin mediate this differentiation. Tissue macrophages have a dual role in the healing process. They are voracious phagocytes and patrol the wound area, ingesting bacteria, devitalised tissue and depleted neutrophils. Macrophages also produce collagenases and elastase to assist them in breaking down devitalised tissues. They are able to regulate proteolytic destruction of tissue in the wound by producing and secreting inhibitors for these enzymes.
Soluble mediators: Macrophages also mediate the transition from the inflammatory phase to the proliferative phase of healing, a role that is as important as their phagocytic role. They release a wide variety of growth factors and cytokines, including TNF-α, TGF-β, PDGFs, IL-1, interleukin 6 (IL-6), insulin-like growth factor-one (IGF-1) and FGF. Some of these soluble mediators recruit and activate fibroblasts, which will synthesise, deposit and organise the new tissue matrix, while others promote angiogenesis.
During the repair phase, the provisional wound matrix is remodelled and replaced with scar tissue, consisting of new collagen fibres, proteoglycans and elastin fibres, which partially restore the structure and function of the tissue. This is accomplished by the migration, proliferation and differentiation of epithelial cells, dermal fibroblasts and vascular endothelial cells from adjacent uninjured tissue and stem cells that originate in the bone marrow and circulate to the wound site.
Fibroblasts in the normal dermis are typically quiescent and sparsely distributed, in contrast to those in the provisional wound matrix and granulation tissue, which are numerous and active. Fibroblasts migrate into the wound in response to soluble cytokines and growth factors, which are initially released from platelets when they degranulate and later by macrophages in the wound. These include PDGF, TGF-β and basic FGF.
The direction of fibroblast movement is determined by the concentration gradient of chemotactic factors and by the alignment of the fibrils in the ECM and provisional matrix. Fibroblasts tend to migrate along these fibrils, as opposed to across them. They begin moving by binding first to matrix components such as collagen, fibronectin, vitronectin and fibrin via their cell-surface integrin receptors. While one end of the fibroblast remains bound to the matrix component, the cell extends a cytoplasmic projection to find another binding site. When the next site is found, the attachment to the original site is broken by proteases secreted by the fibroblast and the cell uses its cytoskeletal network of actin fibres to pull itself forward. These proteases are called matrix metalloproteinases (MMPs) and are essential for the migration of cells through the ECM.
The most important of these MMPs are collagenase (MMP-1), which cuts intact collagen at a single site; gelatinases (MMP-2 and MMP-9), which degrade partially denatured collagen (gelatin); and stromelysin (MMP-3), which degrades multiple protein substrates in the ECM. In addition, MMPs remove collagen and other ECM components that were denatured during the injury. This is important because the collagen molecules must interact very specifically with each other to properly form a collagen fibril (Figure 2). Partially degraded collagen molecules will not bind (interact) properly with new collagen molecules synthesised during scar formation, resulting in disorganised, weak ECM, so the degraded collagen molecules must be removed by controlled action of the MMPs. This is like remodelling a brick wall. When a hole is knocked in a brick wall to permit a new doorway or window, the end of the rows of bricks must be carefully prepared to enable the correct alignment of new bricks that are added to create the correct structure. MMPs ‘chew back’ the denatured matrix to reach intact functional matrix. However, this process must be carefully controlled by tissue inhibitors of metalloproteinases (TIMPs) to prevent the MMPs from degrading intact, functional matrix.
Fibrin peptides that have been degraded by proteolytic enzymes can increase vascular permeability and induce angiogenesis. Peptides of fibronectin, collagen and elastin can stimulate cell migration, influence cell proliferation and lead to arteriolar vasodilation[11],[12],[13]. Thus, the controlled actions of proteases on ECM components play a key role in regulating angiogenesis and many other aspects of normal wound healing, including the migration of cells into the wound.
The process of angiogenesis is stimulated by local factors in the wound microenvironment, including low oxygen tension, low pH and high lactate levels[14]. Several growth factors, including bFGF, TGF-β and vascular endothelial growth factor (VEGF), are also potent angiogenic signals for endothelial cells. It is now recognised that oxygen levels in the tissues directly regulate angiogenesis by interacting with oxygen-sensing proteins that regulate the transcription of angiogenic and anti-angiogenic genes. For example, the synthesis of VEGF by capillary endothelial cells is increased by hypoxia through the activation of the recently identified transcription factor hypoxia-inducible factor (HIF), which binds oxygen[15]. When oxygen levels surrounding capillary endothelial cells drop, levels of HIF increase inside the cells. HIF-1 binds to specific DNA sequences and stimulates the transcription of specific genes, such as that encoding VEGF, that promote angiogenesis. The regulation of angiogenesis involves both stimulatory factors, such as VEGF, and anti-angiogenic factors, such as angiostatin, endostatin, thrombospondin and pigment epithelium-derived factor.
The cells that form the new capillaries were assumed to arise from pre-existing capillary endothelial cells, but it is clear from animal experiments that angiogenic factors cause endothelial cells adjacent to an ischaemic site to begin to migrate into the matrix and proliferate, forming new buds or sprouts. However, recent data suggest that some of the cells that form new capillaries actually come from bone marrow stem cells called haemangioblasts that circulate in the bloodstream[16]. Furthermore, these bone marrow-derived cells have been found to contribute to both collagen type I and III, compared with the resident cell population that only transcribes type I collagen[17]. Angiogenic factors are released as a result of either injury or ischaemia, causing haemangioblasts to migrate into the ECM and differentiate into capillary endothelial cells. The migration of these cells into the matrix requires the local secretion of proteolytic enzymes, especially MMPs. As the tip of a newly formed endothelial sprout comes into contact with another, a cleft develops; this subsequently becomes the lumen of the evolving vessel and a complete new vascular loop is formed. This process continues until the capillary system is sufficiently repaired and tissue oxygenation and metabolic needs are met. It is these new capillary loops embedded in a loosely deposited collagen matrix that give granulation tissue its characteristic uneven or granular appearance.
After the fibroblasts have migrated into the provisional wound matrix, they proliferate and begin to synthesise new collagen, elastin, proteoglycans and other components that comprise granulation tissue. PDGF and TGF-β are two of the important growth factors that regulate the expression of ECM genes and proteases in fibroblasts. Recent data indicate that a new growth factor, named connective tissue growth factor, mediates many of the effects of TGF-β on the synthesis of ECM[18].
This is the final phase of wound healing. The maturation of granulation tissue involves a reduction in the number of capillaries as smaller vessels are aggregated into larger ones, and a decrease in the amount of GAGs and proteoglycans. Cell density and metabolic activity in the granulation tissue decrease during maturation. Changes also occur in the type, amount and organisation of collagen, leading to an enhancement of the tensile strength of the tissues. Initially, type III collagen is synthesised at high levels, but this is replaced by type I collagen, the dominant fibrillar collagen in skin. The tensile strength of a newly epithelialised skin wound is only about 25% of normal tissue[19]. Healed tissue is therefore never as strong as uninjured tissue. Tissue tensile strength is enhanced primarily by the reorganisation of collagen fibres, which are deposited randomly during granulation, and by increased covalent cross-linking of collagen molecules by the enzyme lysyl oxidase, which is secreted into the ECM by fibroblasts. Over several months or more, changes in collagen organisation in the repaired tissue will slowly increase the tensile strength to a maximum of about 80% of normal tissue.
Remodelling of the ECM proteins occurs through the actions of several different classes of proteolytic enzymes produced by cells in the wound bed at different times during the healing process. Two of the most important families are MMPs and serine proteases. Specific MMP proteases that are necessary for wound healing are the collagenases, which degrade intact fibrillar collagen molecules; the gelatinases, which degrade damaged fibrillar collagen molecules; and the stromelysins, which degrade proteoglycans. An important serine protease is neutrophil elastase, which can degrade almost all types of protein molecules. Under normal conditions, the destructive actions of proteolytic enzymes are carefully regulated by specific enzyme inhibitors, which are produced by cells in the wound bed. The TIMPs specifically inhibit MMPs.
All chronic wounds begin as acute wounds with a fibrin clot, but instead of progressing through the four phases of healing they become ‘stuck’ in a prolonged inflammatory phase. It has been proposed that this lengthened inflammatory phase causes increased levels of proteases such as MMPs, elastase, plasmin and thrombin, which destroy components of the ECM and damage the growth factors and their receptors that are essential for healing[20]. Other factors that may contribute to the failure of some wounds to heal include elevated levels of oxygen free radicals, which chemically alter these essential components[21],[22].
Non-healing chronic wounds: Herrick and colleagues[23] investigated sequential changes in the location and levels of key proteins of the ECM, such as fibronectin, during the healing of chronic venous leg ulcers. They examined biopsies of the ulcer base, margin and surrounding skin before and after treatment with compression therapy and observed the following:
Numerous small and medium-sized blood vessels were surrounded by prominent ‘fibrin cuffs’ containing concentric layered structures rich in fibrin, collagen and laminin
The chronic ulcer base was covered with a layer of fibrinous exudate of variable thickness that contained numerous polymorphonuclear leukocytes
Extravasated red blood cells, small perivascular deposits of haemosiderin and macrophages were present in the ulcer margin, base and surrounding skin
Fibronectin staining markedly decreased at the base of the ulcer margins and was virtually absent from the central ulcer base and fibrinous exudates, with the exception of the fibrin cuffs around the blood vessels. Immunostaining for fibronectin showed a diffuse pattern in the ECM of the surrounding skin, with a punctate staining pattern at the epidermal-dermal junction (Figure 4)
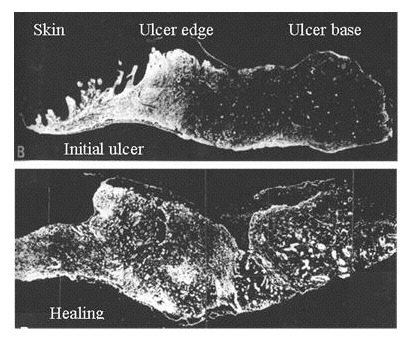
Collagen I and III were evident throughout the ECM of the surrounding skin. Within the ulcer base, bundles of collagen III were visible, lying parallel to the surface and differentiating between the fibrinous exudate and the surface of the granulation tissue. Collagen I staining was less evident in the ulcer base but was present in the basement membrane zone at the ulcer edge.
Healing chronic wounds: After two weeks of compression bandaging therapy, the following important histological changes were apparent in the venous leg ulcers:
The epidermis of the surrounding skin was thicker than normal skin and highly keratinised
The fibrinous exudate was thinner on the ulcer base and regenerating epidermis was visible
Newly formed blood vessels and granulation tissue of variable maturity were present in the ulcer base and margins, and there was a marked increase in the number of polymorphonuclear leukocytes infiltrating the tissue. Macrophages had also increased in number
Blood vessels cuffed with fibrin, laminin, fibronectin, collagen and tenascin were less abundant in both the ulcer edge and base
Some red blood cell extravasation persisted, but there was a striking increase in the amount of haemosiderin deposited in the surrounding tissue
The intensity and distribution of fibronectin staining throughout the ulcer base and edge was dramatically increased compared with the initial biopsy, and collagen III immunostaining was more uniform.
As healing progressed, epithelium covered the edges of the ulcers but was often detached from the underlying dermis. Reduced haemosiderin and focal red blood cell extravasation were observed in the surrounding skin and the ulcer margin. Angiogenesis was pronounced, with capillaries being surrounded by a thin layer of laminin consistent with the thickness of a normal basement membrane, and thicker fibrin cuffs were absent. A fine fibre matrix of fibronectin and collagen was present.
After complete epithelialisation, the pattern of fibronectin and collagen staining of the former ulcer base was indistinguishable from the adjacent surrounding tissue. However, the regenerated epidermis often became detached, suggesting weak attachment of the epidermal cells to the basement membrane. This histological study provides important insight into the molecular, cellular and structural abnormalities that are typical of chronic venous leg ulcers and the progression of changes that occur during healing.
Other studies have compared the differences in wound fluid and ECM in acute wounds and chronic diabetic and venous ulcers[24],[25]. The predominant types of inflammatory cells in early wound healing are granulocytes, lymphocytes and macrophages. The main function of granulocytes in wounds is to eliminate contaminating bacteria. The functions of lymphocytes in acute wound healing are less well established but they probably play a key role in activating macrophages, which are important in regulating the transition between wound inflammation and proliferation. Loots and colleagues[24] found that chronic diabetic or venous ulcers contained significantly higher numbers of granulocytes than acute healing wounds on days five to 28 post-wounding. Levels of B cells, monocytes and macrophages were also significantly higher in chronic wounds than in acute wounds, especially 28 days after injury when the inflammatory response is generally decreasing in healing wounds. By contrast, the numbers of helper T cells were lower in chronic wounds than in acute wounds.
The location and levels of proteoglycans in normal skin, acute wounds and chronic wounds is just beginning to be investigated. An analysis of proteoglycans in venous leg ulcers and normal skin found some differences[25]. In general, the proteoglycans in normal skin tended to be located at the plasma membrane surface of keratinocytes, while in venous ulcers the proteoglycans were mainly located in the keratinocyte cytoplasm. As some proteoglycan molecules bind growth factors and are necessary for their interaction with receptors, alterations in the location and levels of proteoglycans could influence growth factor systems in chronic wounds, justifying further investigations[3].
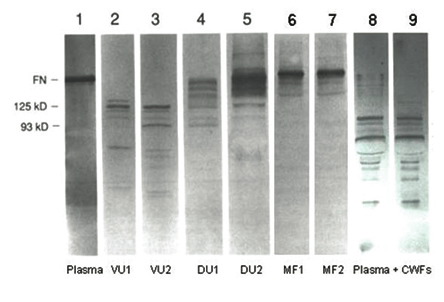
Studies examining the molecular profiles of ECM in chronic wounds produced data that generally corroborated the histological changes observed in the later studies already discussed[23],[24],[25]. One of the first reports to analyse the molecular status of ECM molecules in chronic wounds assessed the stability of fibronectin and vitronectin in fluids collected from chronic wounds[26] (Figure 5). The results demonstrated that fibronectin in plasma (control) and healing surgical wounds was intact. By contrast, fibronectin was totally degraded in the fluid collected from two venous stasis ulcers and was partially degraded in the exudate collected from two chronic diabetic foot ulcers. Importantly, the addition of intact plasma fibronectin to chronic wound fluids resulted in rapid degradation of the fibronectin. These results indicated that levels of intact soluble fibronectin were high in fluids from acute healing wounds but were dramatically reduced in fluids from some chronic wounds. Similar results were found for vitronectin, with significant degradation of vitronectin observed in chronic wound fluids. These results predated but further support the histochemical data of Herrick and colleagues[23], who found reduced immunostaining of fibronectin in chronic venous ulcers.
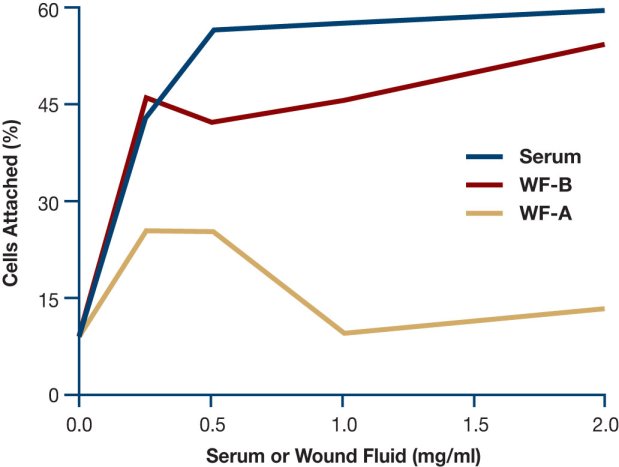
A subsequent study assessed the effect of chronic wound fluids on the attachment of fibroblasts to collagen-coated wells (Figure 6); a high number of human dermal fibroblasts added to medium containing 10% serum attached to the collagen-coated wells. By contrast, reduced levels of cell attachment occurred when fluids from two chronic venous stasis leg wounds were added to the medium[27]. Since fibronectin and vitronectin function as cell attachment and migration factors for many types of cells, degradation of these proteins by high protease activities probably contributes to the delays seen in epidermal resurfacing by keratinocytes, and poor granulation tissue formation by vascular endothelial cells and fibroblasts.
Data on elastin degradation in chronic skin wounds is lacking, but it is known that elastase is responsible for fibronectin degradation in burn wounds[28]. It remains to be determined whether this also indicates that elastin degradation products can be found in chronic wounds. The most likely source of elastase in chronic wounds is from the polymorphonuclear leukocytes[29] or macrophages that are present[30]. Fibroblasts can also express elastase but its action appears to be directed at pre-elastic fibres, with limited activity towards mature dermal elastic fibres[31]. Elastin degradation or its decreased expression leads to the loss of elastolytic properties in skin, for example with ageing or in scar tissue[32].
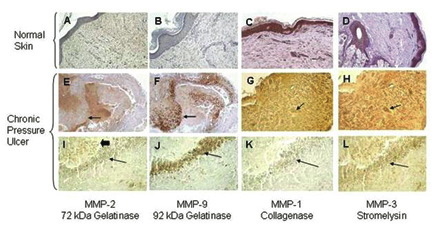
Evidence of collagen degradation in chronic wounds has largely been inferred from data measuring levels of MMPs in fluid obtained from chronic ulcers, rather than by analysing fluids directly for collagen breakdown products. Several studies have reported elevated levels of MMPs[33],[34],[35],[36],[37],[38],[39],[40],[41],[42],[43],[44],[45]. Furthermore, the initial ratio of MMP-9/TIMP-1 in the exudate of chronic pressure ulcers inversely correlated with the healing of pressure ulcers[46]. Analysis of biopsies of chronic pressure ulcers has also shown that, compared with normal skin tissue, MMP levels are highly elevated in pressure ulcers and that there is a strong association with inflammatory cells. For example, as seen in Figure 7, immunostaining for MMP-1, MMP-2, MMP-3, and MMP-9 is very faint or absent in normal skin. In marked contrast, levels of all four MMPs are highly elevated in biopsies of chronic pressure ulcers. Furthermore, immunostaining for MMP-9 and MMP-2 is strongly associated with the inflammatory cells, which in these biopsies are present as a band of cells located in the superficial layers of the ulcer bed.
Other proteases may also play important roles in damaging ECM components or growth factors. The serine protease urokinase plasminogen activator (uPA) is known to activate a proteolytic cascade, resulting in the activation of MMPs. Analysis of fluids and biopsies from acute and chronic wounds found lower levels of active uPA and MMP-9 in acute wounds than in chronic venous leg ulcers[47]]. More importantly, there was a temporal switch from the active to the inactive form of uPA that coincided with a reduction in MMP-9 levels when chronic wounds began to heal. This result reinforces the importance of natural inhibitors of proteases, such as alpha-1 protease inhibitor and TIMPs, in the healing process[48]. The lack of regulation of protease activity in chronic wounds results in degradation of the components of the ECM, leading to the eventual tissue breakdown, ulceration and skin stripping frequently seen in chronic wounds such as venous leg ulcers and pressure ulcers.
These research finding have led to the development of several wound dressings with the aim of inactivating MMPs in chronic wounds. One of these, which consists of oxidised regenerated cellulose and collagen (Promogran®), has been shown to reduce levels of MMP activity and degrade exogenous PDGF in chronic wound fluid in vitro, presumably by acting as a competitive substrate for wound fluid proteases, which spares the PDGF at the expense of degrading the collagen in the dressing[49],[50]. One clinical study suggests that this dressing may improve the healing of some chronic diabetic ulcers[51]. A product that combines collagen with alginate (Fibracol®) has also been developed to absorb wound exudate (alginate component) and reduce MMP protease activity by providing a competitive substrate (collagen component).
A dressing containing a mixture of metal ions and citric acid (DerMax®) is reported to reduce oxygen free radicals and levels of MMP-2 in vitro[52], and small non-randomised studies are now under way to evaluate its use in diabetic foot ulcers and pressure ulcers.
Another new approach in development is to treat wounds topically with a unique protein that normally functions to enhance cell attachment during tooth development[53]. This protein, called amelogenin, is purified from porcine teeth and has been used with substantial success in about 750,000 patients undergoing periodontal procedures. Amelogenin is being evaluated for its ability to promote dermal wound healing based on its ability to provide a temporary ECM protein for cell attachment.
The components of the ECM provide strength, elasticity and compressibility in normal skin. In acute wounds the provisional wound matrix, containing fibrin and fibronectin, plays several key roles, including providing a scaffold to direct cells into the injury as well as stimulating them to proliferate, differentiate and synthesise new ECM. As healing proceeds, the initial ECM of the scar undergoes remodelling and eventually the injured tissue is repaired rather than regenerated because the architecture of the scar never completely reproduces the pre-wound architecture of the skin tissue. In some wounds healing fails to progress through the sequential phases and a chronic wound develops. These wounds are often characterised by increased levels of inflammatory cells that are associated with elevated levels of proteases; these appear to degrade the ECM components, growth factors and receptors that are essential for healing.
Recognising and removing these barriers to healing has led to the concept of wound bed preparation, which emphasises the need to debride non-viable and denatured ECM[54]. The importance of re-establishing a functional ECM in chronic wounds has led to the development of dressings that reduce excessive protease levels, and new technologies are exploring how to temporarily replace the degraded ECM in chronic wounds to facilitate healing.
Clearly, the optimal care and treatment of chronic wounds requires an understanding of the key role ECM plays in normal healing, recognition of how this is altered in chronic wounds and the inclusion of appropriate clinical treatments to re-establish a functional ECM.
Chemokines are small molecules released by cells at the site of injury or infection which bind to receptors on the surface of target cells, giving rise to intracellular signals that stimulate chemotaxis (cell motion towards a chemical gradient). An example of chemotaxis is the migration of leukocytes into the tissue from blood vessels.
Chemotaxis is the locomotion of cells or organisms determined by diffusible substances in their environment. The direction of movement is affected by the concentration gradient of the substance.
Cytokines are small, cell-secreted polypeptides (proteins) that affect the behaviour of cells – for example, the expression of growth factors by macrophages, the migration of polymorphonuclear leukocytes (neutrophils) to the wound site and fibroblast proliferation. Cytokines are released, mainly by polymorphonuclear leukocytes and macrophages, during episodes of inflammation and are crucial to the normal healing process. Examples include tumour necrosis factor alpha (TNF-α) and interleukin-1 beta (IL-1β). Technically, growth factors and chemokines are subsets of cytokines, but the term cytokine usually refers to molecules that primarily target inflammatory cells.
The cytoskeleton supports the cell and gives it its shape. It is composed of microtubules and filaments, which are necessary for cell motility and aid the movement of materials in and out of cells.
Haemosiderin is an insoluble iron-protein molecule composed of ferric oxide which is a source of iron for haemoglobin synthesis and other metabolic processes. It is one form of iron storage within tissues and is visible microscopically with special stains.
Glycosaminoglycans (GAGs) are large polysaccharide chains that are made up of repeating disaccharide units that are negatively charged and hold a large amount of water compared to other ECM molecules. GAGs provide a majority of the resistance to compression in tissues while collagen molecules provide tensile strength (resistance to breaking or tearing). GAGs are typically found bound to the core proteins of proteoglycans, and examples include dermatan sulphate proteoglycan, keratin sulphate proteoglycan and hyaluronic acid.
Glycoprotein is a protein with attached branching carbohydrates. Many membrane proteins are glycoproteins. These may function in cell-to-cell recognition, such as in human blood groups and immune system responses, as well as in resisting the compression of cells. Examples include cell-bound fibronectin.
Growth factors are small proteins that are synthesised by cells at the site of a wound or released from granules in platelets during clotting which stimulate proliferation, migration and differentiation of the wound cells, especially keratinocytes, fibroblasts and vascular endothelial cells, which are their main target cells. Growth factors differ from cytokines and chemokines in that their typical target cells are not inflammatory cells. Examples include epidermal growth factor (EGF), platelet derived growth factor (PDGF) and vascular endothelial cell growth factor (VEGF).
Cells respond to signals received from molecules outside the cell that bind to integral membrane proteins that transmit the signals into the cell. They also possess cell-surface chemicals (receptor sites) that allow them to communicate with and influence other cells with which they are in direct physical contact. These receptor sites on the surface of a cell can also help them to adhere to the ECM.
Matrix metalloproteinases are proteolytic enzymes, such as collagenases and gelatinases, which break down collagens and other matrix molecules and help to remodel the ECM. MMPs are members of the metalloproteinase super family of proteases, which includes diverse proteases such as TNF-α converting enzyme (TACE) and angiotensin converting enzyme (ACE).
Proteoglycans are protein and polysaccharide complexes with a high molecular weight that are characteristic of the structural tissues of vertebrates, such as bone and cartilage. They are also present on cell surfaces and play an important part in determining the visco-elastic properties of joints and other structures that are subject to mechanical deformation.
1. Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Watson JD. Cell junctions, cell adhesion, and the extracellular matrix. In: Molecular Biology of the Cell. New York, NY: Garland Science, 2002; 1065-125.
2. Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J. Integrating cells into tissues. In: Molecular Cell Biology. New York, NY: WH Freeman & Co, 2000; 968-1002.
3. Pellegrini L, Burke DF, von Delft F, Mulloy B, Blundell TL. Crystal structure of fibroblast growth factor receptor ectodomain bound to ligand and heparin. Nature 2000; 407(6807): 1029-34.
4. Ruoslahti E, Yamaguchi Y. Proteoglycans as modulators of growth factor activities. Cell 1991; 64(5): 867-9.
5. Kainulainen V, Wang H, Schick C, Bernfield M. Syndecans, heparan sulfate proteoglycans, maintain the proteolytic balance of acute wound fluids. J Biol Chem 1998; 273(19): 11563-9.
6. Bennett NT, Schultz GS. Growth factors and wound healing: Part II. Role in normal and chronic wound healing. Am J Surg 1993; 166(1): 74-81.
7. Bennett NT, Schultz GS. Growth factors and wound healing: biochemical properties of growth factors and their receptors. Am J Surg 1993; 165(6): 728-37.
8. Lawrence WT. Physiology of the acute wound. Clin Plast Surg 1998; 25(3): 321-40.
9. Greiling D, Clark RA. Fibronectin provides a conduit for fibroblast transmigration from collagenous stroma into fibrin clot provisional matrix. J Cell Sci 1997; 110(Pt 7): 861-70.
10. Geer DJ, Andreadis ST. A novel role of fibrin in epidermal healing: plasminogen-mediated migration and selective detachment of differentiated keratinocytes. J Invest Dermatol 2003; 121(5): 1210-6.
11. Boudreau N, Bissell MJ. Extracellular matrix signaling: integration of form and function in normal and malignant cells. Curr Opin Cell Biol 1998; 10(5): 640-6.
12. Ruoslahti E. Fibronectin and its integrin receptors in cancer. Adv Cancer Res 1999; 76: 1-20.
13. Lukashev ME, Werb Z. ECM signalling: orchestrating cell behaviour and misbehaviour. Trends Cell Biol 1998; 8(11): 437-41.
14. Bhushan M, Young HS, Brenchley PE, Griffiths CE. Recent advances in cutaneous angiogenesis. Br J Dermatol 2002; 147(3): 418-25.
15. Semenza GL. HIF-1 and tumor progression: pathophysiology and therapeutics. Trends Mol Med 2002; 8(4 Suppl): S62-7.
16. Grant MB, May WS, Caballero S, Brown GA, Guthrie SM, Mames RN, et al. Adult hematopoietic stem cells provide functional hemangioblast activity during retinal neovascularization. Nat Med 2002; 8(6): 607-12.
17. Fathke C, Wilson L, Hutter J, Kapoor V, Smith A, Hocking A, et al. Contribution of bone marrow-derived cells to skin: collagen deposition and wound repair. Stem Cells 2004; 22(5): 812-22.
18. Duncan MR, Frazier KS, Abramson S, Williams S, Klapper H, Huang X, et al. Connective tissue growth factor mediates transforming growth factor beta-induced collagen synthesis: down-regulation by cAMP. FASEB J 1999; 13(13): 1774-86.
19. Levenson SM, Geever EF, Crowley LV, Dates JF, Berard CW, Rosen H. The healing of rat skin wounds. Ann Surg 1965; 161: 293-308.
20. Mast BA, Schultz GS. Interactions of cytokines, growth factors, and proteases in acute and chronic wounds. Wound Repair Regen 1996; 4(4): 411-20.
21. James TJ, Hughes MA, Cherry GW, Taylor RP. Evidence of oxidative stress in chronic venous ulcers. Wound Repair Regen 2003; 11(3): 172-6.
22. Moseley R, Hilton JR, Waddington RJ, Harding KG, Stephens P, Thomas DW. Comparison of oxidative stress biomarker profiles between acute and chronic wound environments. Wound Repair Regen 2004; 12(4): 419-29.
23. Herrick SE, Sloan P, McGurk M, Freak L, McCollum CN, Ferguson MW. Sequential changes in histologic pattern and extracellular matrix deposition during the healing of chronic venous ulcers. Am J Pathol 1992; 141(5): 1085-95.
24. Loots MA, Lamme EN, Zeegelaar J, Mekkes JR, Bos JD, Middelkoop E. Differences in cellular infiltrate and extracellular matrix of chronic diabetic and venous ulcers versus acute wounds. J Invest Dermatol 1998; 111(5): 850-7.
25. Lundqvist K, Schmidtchen A. Immunohistochemical studies on proteoglycan expression in normal skin and chronic ulcers. Br J Dermatol 2001; 144(2): 254-9.
26. Wysocki AB, Grinnell F. Fibronectin profiles in normal and chronic wound fluid. Lab Invest 1990; 63(6): 825-31.
27. Grinnell F, Ho CH, Wysocki A. Degradation of fibronectin and vitronectin in chronic wound fluid: analysis by cell blotting, immunoblotting, and cell adhesion assays. J Invest Dermatol 1992; 98(4): 410-6.
28. Grinnell F, Zhu M. Identification of neutrophil elastase as the proteinase in burn wound fluid responsible for degradation of fibronectin. J Invest Dermatol 1994; 103(2): 155-61.
29. Diegelmann RF. Excessive neutrophils characterize chronic pressure ulcers. Wound Repair Regen 2003; 11(6): 490-5.
30. Chapman HA, Munger JS, Shi GP. The role of thiol proteases in tissue injury and remodeling. Am J Respir Crit Care Med 1994; 150(6 Pt 2): S155-9.
31. Godeau G, Hornebeck W. Morphometric analysis of the degradation of human skin elastic fibres by human leukocyte elastase (EC 3-4-21-37) and human skin fibroblast elastase (EC 3-4-24). Pathol Biol (Paris) 1988; 36(9): 1133-8.
32. Kamath NV, Ormsby A, Bergfeld WF, House NS. A light microscopic and immunohistochemical evaluation of scars. J Cutan Pathol 2002; 29(1): 27-32.
33. Wysocki AB, Staiano-Coico L, Grinnell F. Wound fluid from chronic leg ulcers contains elevated levels of metalloproteinases MMP-2 and MMP-9. J Invest Dermatol 1993; 101(1): 64-8.
34. Yager DR, Nwomeh BC. The proteolytic environment of chronic wounds. Wound Repair Regen 1999; 7(6): 433-41.
35. Yager DR, Zhang LY, Liang HX, Diegelmann RF, Cohen IK. Wound fluids from human pressure ulcers contain elevated matrix metalloproteinase levels and activity compared to surgical wound fluids. J Invest Dermatol 1996; 107(5): 743-8.
36. Yager DR, Chen SM, Ward SI, Olutoye OO, Diegelmann RF, Cohen K. Ability of chronic wound fluids to degrade peptide growth factors is associated with increased levels of elastase activity and diminished levels of proteinase inhibitors. Wound Repair Regen 1997; 5(1): 23-32.
37. Trengove NJ, Stacey MC, MacAuley S, Bennett N, Gibson J, Burslem F, et al. Analysis of the acute and chronic wound environments: the role of proteases and their inhibitors. Wound Repair Regen 1999; 7(6): 442-52.
38. Rogers AA, Burnett S, Moore JC, Shakespeare PG, Chen WYJ. Involvement of proteolytic enzymes - plasminogen activators and matrix metalloproteinases - in the pathophysiology of pressure ulcers. Wound Repair Regen 1995; 3(3): 273-83.
39. Bullen EC, Longaker MT, Updike DL, Benton R, Ladin D, Hou Z, et al. Tissue inhibitor of metalloproteinases-1 is decreased and activated gelatinases are increased in chronic wounds. J Invest Dermatol 1995; 104(2): 236-40.
40. Barone EJ, Yager DR, Pozez AL, Olutoye OO, Crossland MC, Diegelmann RF, et al. Interleukin-1alpha and collagenase activity are elevated in chronic wounds. Plast Reconstr Surg 1998; 102(4): 1023-7; discussion 1028-9.
41. Tarnuzzer RW, Schultz GS. Biochemical analysis of acute and chronic wound environments. Wound Repair Regen 1996; 4(3): 321-25.
42. Harris IR, Yee KC, Walters CE, Cunliffe WJ, Kearney JN, Wood EJ, et al. Cytokine and protease levels in healing and non-healing chronic venous leg ulcers. Exp Dermatol 1995; 4(6): 342-9.
43. Weckroth M, Vaheri A, Lauharanta J, Sorsa T, Konttinen YT. Matrix metalloproteinases, gelatinase and collagenase, in chronic leg ulcers. J Invest Dermatol 1996; 106(5): 1119-24.
44. Nwomeh BC, Liang HX, Cohen IK, Yager DR. MMP-8 is the predominant collagenase in healing wounds and nonhealing ulcers. J Surg Res 1999; 81(2): 189-95.
45. Lobmann R, Ambrosch A, Schultz G, Waldmann K, Schiweck S, Lehnert H. Expression of matrix-metalloproteinases and their inhibitors in the wounds of diabetic and non-diabetic patients. Diabetologia 2002; 45(7): 1011-6.
46. Ladwig GP, Robson MC, Liu R, Kuhn MA, Muir DF, Schultz GS. Ratios of activated matrix metalloproteinase-9 to tissue inhibitor of matrix metalloproteinase-1 in wound fluids are inversely correlated with healing of pressure ulcers. Wound Repair Regen 2002; 10(1): 26-37.
47. Wysocki AB, Kusakabe AO, Chang S, Tuan TL. Temporal expression of urokinase plasminogen activator, plasminogen activator inhibitor and gelatinase-B in chronic wound fluid switches from a chronic to acute wound profile with progression to healing. Wound Repair Regen 1999; 7(3): 154-65.
48. Rao CN, Ladin DA, Liu YY, Chilukuri K, Hou ZZ, Woodley DT. Alpha 1-antitrypsin is degraded and non-functional in chronic wounds but intact and functional in acute wounds: the inhibitor protects fibronectin from degradation by chronic wound fluid enzymes. J Invest Dermatol 1995; 105(4): 572-8.
49. Cullen B, Smith R, McCulloch E, Silcock D, Morrison L. Mechanism of action of PROMOGRAN, a protease modulating matrix, for the treatment of diabetic foot ulcers. Wound Repair Regen 2002; 10(1): 16-25.
50. Cullen B, Watt PW, Lundqvist C, Silcock D, Schmidt RJ, Bogan D, et al. The role of oxidised regenerated cellulose/collagen in chronic wound repair and its potential mechanism of action. Int J Biochem Cell Biol 2002; 34(12): 1544-56.
51. Veves A, Sheehan P, Pham HT. A randomized, controlled trial of Promogran (a collagen/oxidized regenerated cellulose dressing) vs standard treatment in the management of diabetic foot ulcers. Arch Surg 2002; 137(7): 822-7.
52. van den Berg AJ, Halkes SB, van Ufford HC, Hoekstra MJ, Beukelman CJ. A novel formulation of metal ions and citric acid reduces reactive oxygen species in vitro. J Wound Care 2003; 12(10): 413-8.
53. Hoang AM, Klebe RJ, Steffensen B, Ryu OH, Simmer JP, Cochran DL. Amelogenin is a cell adhesion protein. J Dent Res 2002; 81(7): 497-500.
54. Schultz GS, Sibbald RG, Falanga V, Ayello EA, Dowsett C, Harding K, et al. Wound bed preparation: a systematic approach to wound management. Wound Repair Regen 2003; 11(Suppl 1): S1-S28.