
|
Author(s)
Stuart Enoch
Patricia Price
|
Contents
|
|
Published:
Aug 2004
Last updated: Aug 2004 Revision: 1.0 |
Keywords: acute wounds; chronic wounds; pathophysiology of healing; wound infection; ageing; assessment; outcome measures.
Chronic wound healing does not follow the same pattern as that of acute wounds.
A significant number of chronic or non-healing wounds are more prevalent in older people so the effects of ageing on the healing process need to be understood and taken into account when assessing older people.
There is an urgent need to develop meaningful alternative endpoints, as opposed to complete wound closure, for use when assessing patients with chronic or non-healing wounds.
Acute wound healing is a dynamic process involving the coordinated actions of both resident and migratory cell populations within the extracellular matrix environment leading to the repair of injured tissues. In contrast to this some wounds fail to heal in a timely and orderly manner, resulting in chronic non-healing wounds. In addition, chronic wounds are more prevalent in older people due to the altered molecular and cellular characteristics of the aged skin and various associated co-morbidities. This article discusses in detail the cellular, molecular and biochemical differences in healing between acute and chronic wounds, and outlines the effects of ageing on the healing process.
Skin, the largest organ in the human body, plays a crucial role in the sustenance of life through the regulation of water and electrolyte balance, thermoregulation, and by acting as a barrier to external noxious agents including micro-organisms. When this barrier is disrupted due to any cause - ulcers, burns, neoplasm or trauma - these functions are no longer adequately performed. It is therefore vital to restore its integrity as soon as possible.
A wound is defined as a break in the epithelial integrity of the skin. However, the disruption could be deeper, extending to the dermis, subcutaneous fat, fascia, muscle or even the bone. Normal wound healing involves a complex and dynamic but superbly orchestrated series of events leading to the repair of injured tissues. A completely healed wound, usually seen after simple injury, is defined as one that has returned to its normal anatomical structure, function and appearance within a reasonable period of time. It is also defined as one that has attained complete skin closure without drainage or dressing requirements. In contrast to these some wounds fail to heal in a timely and orderly manner, resulting in chronic, non-healing wounds. Despite advances in molecular biology, the development of various tissue-engineered skin substitutes and growth factors, and a range of other therapeutic options, chronic ulceration remains a significant problem in our society.
Chronic wounds result from various causes, including venous (chronic venous leg ulcers or CVLUs), arterial, neuropathic, pressure, vasculitis and burns. Although chronic ulceration can affect any anatomical region, the most common site is the lower limb and the estimated prevalence of active leg ulceration in Europe is at least 0.1-0.3 percent [1], [2], [3]. Ulcers secondary to venous hypertension and venous insufficiency accounts for nearly 70 per cent of all leg ulcers [4], with diabetes and arterial disease contributing towards a significant proportion of the rest. As all these conditions are more prevalent in older people they are more susceptible to leg ulcers. In addition, the increased occurrence and longevity of these ulcers are further compounded by the detrimental effects ageing has on the skin and the wound healing process.
This article outlines the cellular, molecular and biochemical differences between the acute and the chronic wound environment. The effects of ageing on healing are also reviewed as a significant number of non-healing chronic wounds are prevalent in older people.
Healing in acute wounds occurs as a sequential cascade of overlapping processes that requires the coordinated completion of a variety of cellular activities including phagocytosis, chemotaxis, mitogenesis, collagen synthesis and the synthesis of other matrix components. These activities do not occur in a haphazard manner but rather in a carefully regulated and systematic cascade that correlates with the appearance of different cell types in the wound during various stages of the healing process (Figure 1). These processes, which are triggered by tissue injury, involve the four overlapping but well-defined phases of haemostasis, inflammation, proliferation and remodelling [5]. The regulation of these events is multifactorial and is discussed below.
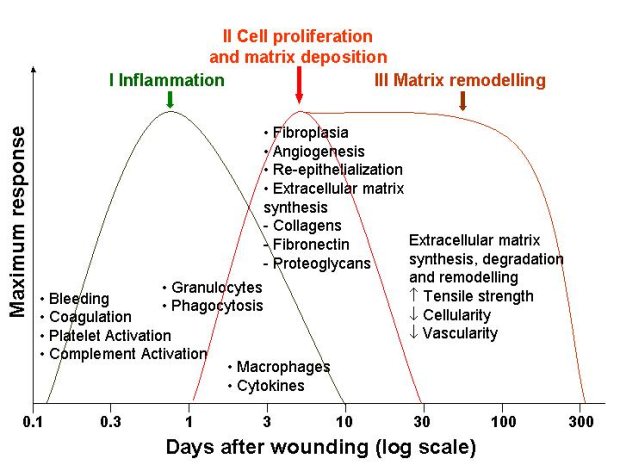
Tissue injury is characterised by microvascular injury and therefore extravasation of blood into the wound. Injured vessels constrict rapidly and the coagulation cascade is activated to limit blood loss, leading to clot formation and platelet aggregation. The clot, comprising of fibrin, fibronectin, vitronectin, von Willebrand factor and thrombospondin, provides the provisional matrix for cellular migration [6], [7]. The platelets trapped in the clot are essential for haemostasis as well as for a normal inflammatory response. The alpha granules of the platelets contain growth factors, including platelet-derived growth factor (PDGF), insulin-like growth factor-1 (IGF-1), epidermal growth factor (EGF), and transforming growth factor-beta (TGF-β). These proteins initiate the wound healing cascade by attracting and activating fibroblasts, endothelial cells and macrophages. The platelets also contain dense bodies that store vasoactive amines such as serotonin that increase microvascular permeability. This leads to the exudation of fluid into the extravascular space and results in tissue oedema, although this feature is more prominent during the inflammatory phase.
The next phase of healing is inflammation, which begins with the activation of complement and the initiation of the classical molecular cascade that leads to infiltration of the wound with granulocytes or polymorphonuclear leucocytes (PMNLs). These cells are attracted to the wound site within 24 to 48 hours of injury by a number of agents, including complement components such as C5a, platelets, formyl-methionyl peptide products from bacteria and TGF-β.
Within a short time, the PMNLs begin to adhere to the endothelial cells in the adjacent blood vessels through a process called margination and start to move through the vessel wall, a process known as diapedesis. Once in the wound environment they phagocytose bacteria and other foreign particles, killing them by releasing degrading enzymes and oxygen-derived free radical species. PMNL activity usually ceases within a few days of wounding once contaminating bacteria have been cleared. Redundant cells are cleared away from the wound by extrusion to the wound surface as slough or phagocytosis by macrophages. The main function of PMNLs is to prevent infection so they contribute little to the normal wound healing process beyond this stage.
Blood monocytes undergo a phenotypic change on arrival at the wound site to become tissue macrophages. Monocytes are attracted to the wound by a variety of chemoattractants, including complement, clotting components, immunoglobulin G (IgG) fragments, collagen and elastin breakdown products, and cytokines such as leukotriene B4, platelet factor IV, PDGF and TGF-β. Macrophages are the most important cells present in the later stages of the inflammatory process (48-72 hours) and appear to act as the key regulatory cells for repair. They release further cytokines and growth factors into the wound, recruiting fibroblasts, keratinocytes and endothelial cells to repair the damaged blood vessels [8]. Macrophages are also capable of releasing proteolytic enzymes such as collagenase that can debride tissue. The depletion of circulating monocytes and tissue macrophages causes severe alterations in wound healing, leading to poor wound debridement, delayed fibroblast proliferation, inadequate angiogenesis and poor fibrosis. Additional growth factors such as transforming growth factor-alpha (TGF-α), heparin-binding epidermal growth factor (HB-EGF), and basic fibroblast growth factor (bFGF) are secreted by the PMNLs and macrophages, which further stimulate the inflammatory response.
The lymphocyte is the last cell type to enter the wound during the inflammatory phase (>72 hours after wounding) and may be attracted by interleukin-1 (IL-1), IgG and complement products. IL-1 is believed to play a key role in the regulation of collagenase, indicating that the lymphocyte may be involved in collagen and extracellular matrix (ECM) remodelling. The role of lymphocytes in wound healing, however, has not been clearly defined.
The proliferative phase starts at about day three and lasts for two weeks after wounding. It is characterised by the replacement of the provisional fibrin/fibronectin matrix with newly formed granulation tissue.
Fibroblast migration: Fibroblasts and myofibroblasts appear in the wound between 2 and 4 days after wounding. Following injury they are stimulated to migrate into the wound defect, proliferate and produce the matrix proteins fibronectin, hyaluronan (HA) and later collagen and proteoglycans. Fibroblasts are attracted by a number of factors including PDGF and TGF-β [9]. Once within the wound environment, fibroblasts proliferate and start to construct the new ECM, which is essential for the repair process and supports further ingrowth of cells. Interactions between the fibroblasts and the ECM itself help to determine the synthesis and remodelling of the matrix [10].
Collagen synthesis: Collagens, which are synthesised by fibroblasts, provide strength and integrity for all tissues in the body and therefore play a particularly vital role in wound repair. Collagens are a key component of all phases of wound healing. Immediately after injury, exposed collagen comes into contact with blood, promoting platelet aggregation and activating chemotactic factors involved in the response to injury. Later collagen becomes the foundation of the wound ECM. Invading fibroblasts synthesise and secrete types I and III collagen to form the new matrix.
Angiogenesis: The process of forming new blood vessels occurs concurrently during all stages of the healing process. TGF-β and PDGF, secreted by the platelets during the haemostatic phase, attract macrophages and granulocytes and promote angiogenesis. The macrophages, in particular, play a key role in angiogenesis by releasing a number of other angiogenic substances including tumour necrosis factor-α; and bFGF. Angiogenic capillary sprouts invade the fibrin/fibronectin-rich wound clot and organise into a microvascular network throughout the granulation tissue within a few days [11]. As collagen accumulates in the granulation tissue to produce scar tissue the density of blood vessels diminishes. Disturbance of this dynamic process may influence the development of chronic wounds [12].
Granulation tissue formation: Granulation tissue is so called because of the pink granular appearance of numerous capillaries that invade the wound stroma. Each 'granule' contains a loop of capillaries and therefore bleeds easily if traumatised. Granulation tissue is made up mainly of proliferating fibroblasts, capillaries and tissue macrophages in a matrix of collagen, glycosaminoglycans (GAGs) including HA, and the glycoproteins fibronectin and tenascin [13], [14]. Granulation tissue formation is evident as early as 48 hours after wounding and by 96 hours fibroblasts become the predominant cell type in this tissue [15].
Epithelialisation: A single layer of epidermal cells start to migrate from the wound edges within a few hours of wounding to form a delicate covering over the raw area exposed by the loss of epidermis, a process known as epiboly. From about 12 hours after wounding there is a marked increase in mitotic activity in the basal cells from the wound edges or around skin appendages. These cells loosen their normally firm attachments to the underlying dermis, allowing them to migrate in a leap-frog fashion across the provisional matrix [16]. When advancing epithelial cells meet, further movement is halted by contact inhibition and a new basement membrane regenerates. Further epithelial cell growth and differentiation re-establishes the stratified epithelium. The rate of epithelial coverage is increased if the wound does not require debridement, if the basal lamina is intact and if the wound is kept moist. A dry eschar (scab) slows the rate of epithelialisation. Several growth factors modulate epithelialisation: EGF is a potent stimulator of epithelial mitogenesis and chemotaxis, while other growth factors, such as bFGF and keratinocyte growth factor, also stimulate epithelial proliferation.
Matrix synthesis and the remodelling phase are initiated concurrently with the development of granulation tissue and continue over prolonged periods of time. As the matrix matures, fibronectin and HA are broken down and collagen bundles increase in diameter, corresponding with increasing wound tensile strength [17], [18]. However, these collagen fibres never regain the original strength of normal unwounded skin and only a maximum of 80 percent unwounded skin strength can be achieved [19].
There is ongoing collagen synthesis and breakdown as the ECM is continually remodelled, equilibrating to a steady state about 21 days after wounding. Collagen degradation is achieved by specific matrix matalloproteinases (MMPs) that are produced by many cells at the wound site, including fibroblasts, granulocytes and macrophages. As remodelling of the wound continues, MMP activity decreases and tissue inhibitors of metalloproteinases (TIMPs) activity increases. TGF-β plays an important role in mediating this, underlining the ability of TGF-β to promote matrix accumulation.
Early collagen deposition is highly disorganised but its subsequent organisation is primarily achieved by wound contraction. Wound remodelling occurs when the underlying contractile connective tissue shrinks in size to bring the wound margins closer together. Contraction occurs through the interactions between fibroblasts and the surrounding ECM. These interactions may be influenced by a number of extracellular factors including TGF-β, PDGF and FGF [10]. With time the density of macrophages and fibroblasts is reduced by apoptosis triggered by unknown sources [20]. However, it has been suggested that apoptosis may be signalled by the withdrawal of cytokines as the wound heals, although many other theories exist including myofibroblast differentiation itself signalling apoptosis and the release of certain factors following re-epithelialisation [21]. With continued remodelling the outgrowth of capillaries is halted, blood flow to the area is reduced and metabolic activity in the area declines. An acellular, avascular scar is the final result of an acute wound healing process.
A chronic wound is defined as one in which the normal process of healing is disrupted at one or more points in the phases of haemostasis, inflammation, proliferation and remodelling [22]. In most chronic wounds, however, the healing process is thought to be 'stuck' in the inflammatory or proliferative phases. As growth factors, cytokines, proteases, and cellular and extracellular elements all play important roles in different stages of the healing process, alterations in one or more of these components could account for the impaired healing observed in chronic wounds. In addition, oxidative damage by free radicals or condition-specific factors such as neuropathy in diabetes or ischaemia in peripheral vascular disease may lead to the non-healing nature of chronic wounds. Healing may also be impeded by the accumulation of necrotic tissue or slough, a feature of chronic wounds.
Alterations in the activities of proteases and their inhibitors have been implicated in the failure of chronic wounds to heal. In the phases of normal wound healing the production and activity of proteases are tightly regulated but this regulation appears to be disrupted in chronic wounds. Levels of various MMPs and serine proteases are markedly increased in fluids from chronic wounds; levels of MMP-1 (collagenase), 2 (gelatinase A) and 9 (gelatinase B) have been shown to be elevated in fluid derived from pressure ulcers and CVLUs compared with acute mastectomy wounds [23], [24]. Similarly, during granulation tissue formation in chronic pressure ulcers, levels of MMPs have been shown to be decreased while levels of their inhibitors, TIMPs, have been found to be elevated [25]. Other proteases, such as neutrophil elastase, have also been observed to be significantly higher in chronic wounds [26]. Elevated levels of serine proteases degrade fibronectin, an essential protein involved in the remodelling of the ECM, and in vitro studies suggest that certain growth factors are also degraded by proteases [23], [27]. Other molecular and biochemical changes are summarised in Table 1.
| Elevated | Decreased |
| Collagenolytic activity - Matrix metalloproteinases (MMP) - 1, 8 and 13 | Tissue inhibitor of metalloproteinases (TIMPs) |
| Gelatinases A (MMP 2) and Gelatinase B (MMP 9) | α1 - protease inhibitor |
| Stromelysins - MMP 3, 10 and 11 | α2 - macroglobulin |
| Serine proteases - Urokinase-type plasminogen activator, Cathepsin G, increased neutrophil elastase activity | Increased degradation of:
|
Wound fluid derived from CVLUs is rich in pro-inflammatory cytokines such as TNF-αa and interleukin-1β (IL-1β), and TGF-β1 [28]. In addition, the levels of these cytokines decreases as the chronic wound begin to heal, indicating a significant correlation between non-healing wounds and increased levels of pro-inflammatory cytokines [29]. Chronic wound fluid containing the above cytokines has also been shown to inhibit growth and induce morphological changes in normal skin fibroblasts [30].
The normal inflammatory response seen in acute wound healing is significantly altered in chronic wounds. PMNLs from patients with CVLUs produce more reactive oxygen species than healthy controls [31]. Macrophage activation, essential for the release of cytokines and growth factors to recruit fibroblasts, keratinocytes and endothelial cells, is suppressed in CVLU, leading to an impaired inflammatory response [32]. Likewise, the lymphocyte infiltration in chronic wounds is also altered: Loots and colleagues reported a lower ratio of CD4+/CD8+ T lymphocytes in chronic wounds compared with acute healing wounds [33].
Differences in the morphology and proliferation of chronic wound fibroblasts have also been reported by many authors. Chronic wound fibroblasts have been described as larger and polygonal in shape compared with the compact, spindle shape of normal skin fibroblasts [34], [35]. Fibroblasts isolated from CVLUs [34], [35], [36], distal skin in patients with venous hypertension [36] and pressure ulcers [37] all show decreased proliferation in comparison with patient-matched normal skin fibroblasts.
A failure to re-epithelialise is perhaps the most obvious clinical feature of chronic wounds. This is thought to be due to a failure in migration rather than proliferation of the keratinocytes [38], [39]. Keratinocyte migration is dependent upon various factors including the underlying matrix and the cytokines released by fibroblasts and macrophages into the wound environment. In acute wounds migrating keratinocytes express α5β1 integrin. In chronic wounds, however, there is reduced expression of α5β1, indicating a non-migratory phenotype of the keratinocytes [40]. Induction of TNF-α production in vitro increases the expression of α5β1 by chronic wound keratinocytes, stimulating a migratory phenotype [40].
Chronic wounds in general display reduced mitogenic activity compared to acute wounds. In contrast to acute wound fluid, when chronic wound fluid is added to cultures of fibroblasts, keratinocytes or vascular endothelial cells it fails to stimulate DNA synthesis in these cells [28], [41]. Similarly, fibroblasts in chronic ulcers may not be capable of responding to growth factors such as PDGF and TGF [35], and fibroblasts isolated from CVLUs demonstrate reduced motility or migration compared with fibroblasts from patient-matched normal skin [42]. Some of the cellular features of chronic wounds are summarised in Table 2.
|
The composition and reorganisation of the ECM in chronic wounds may be defective or altered, leading to delayed re-epithelialisation [43]. Herrick et al observed a decrease in the amount of ECM proteins such as fibronectin in biopsies taken from CVLUs compared to surrounding normal skin and that the fibronectin content increased as the ulcer progressed towards healing [43]. Herrick and colleagues also observed that chronic wound fibroblasts synthesised decreased amounts of collagen [44], although other investigators found no differences unless stimulated with TGF-β [45]. In addition, Cook and colleagues [25] observed that, compared to normal skin fibroblasts, patient-matched chronic wound fibroblasts had a reduced ability to contract a fibroblast populated type I collagen lattice model, suggesting a decreased ability to reorganise the ECM environment in vivo.
Oxygen-derived free radicals have been implicated in the development of venous ulcers and their persistence. Scavenging such radicals using antioxidants expedites healing in venous ulcers [46]. Nitric oxide (NO) is known to combine with hydroxyl free radicals to form peroxynitrate, a potent free radical that causes tissue destruction. NO over-expression may be involved directly or indirectly in the pathogenesis and delayed healing of CVLUs through the production of peroxynitrate and its effects on vasculature, inflammation and collagen deposition [47]. In a study of 44 patients with chronic venous disease, Howlander and Smith observed that the total plasma NO levels were elevated in patients with severe skin damage [48]. Similarly, Jude et al found that diabetic patients with recurrent neuropathic and neuroischaemic foot ulcers had significantly higher plasma NO levels compared with patients with non-recurrent foot ulcers [49].
Due to underlying pathogenic abnormalities and the altered biochemical and cellular environment, necrotic tissue and slough tends to accumulate continually in chronic wounds [50]. Necrotic tissue, the result of inadequate local blood supply, contains dead cells and debris that are a consequence of the fragmentation of dying cells. Slough is a yellow fibrinous tissue that consists of fibrin, pus and proteinaceous material. The accumulation of necrotic tissue or slough in a chronic wound promotes bacterial colonisation and prevents complete repair of the wound. Necrotic burden is an all-encompassing term that describes necrotic tissue, slough, excess exudate and high levels of bacteria present in chronic wound environment. If the necrotic burden is allowed to accumulate in a chronic wound it can prolong the inflammatory response, mechanically obstruct the process of wound contraction and impede re-epithelialisation [51].
The existence of bacteria in the wound bed can be divided into four distinct categories based on the induced host response:
Contamination (presence of non-replicating micro-organisms within a wound)
Colonisation (replicating micro-organisms that adhere to the wound surface in the absence of tissue damage)
Local infection or critical colonisation (wound with an increasing bacterial burden, which is an intermediate category between colonisation and invasive infection)
Invasive wound infection (presence of replicating micro-organisms within a wound with a subsequent host response that leads to delayed healing).
However, the relative number of micro-organisms (usually more than 105 per gram of tissue) and their pathogenicity (such as beta haemolytic streptococci, which are highly virulent), in combination with host response and factors such as immunodeficiency, diabetes mellitus and drugs, dictate whether a chronic wound becomes infected or shows signs of delayed healing. The characteristics of bacteria in chronic wounds are summarised in Table 3.
|
In addition to episodes of infection, the continued presence of bacteria in a wound leads to endotoxin production and stimulates the host's immune defences to produce pro-inflammatory mediators such as IL-1, TNF-α, prostaglandin E2 and thromboxane. Although inflammation is part of normal wound healing, the repair process may be prolonged if the inflammation is excessive [52]. In addition, with chronic colonisation bacteria in wounds form biofilms (bacterial colonies embedded in a self-secreted extracellular polysaccharide matrix) that are resistant to the action of host defences and antimicrobial agents, thereby contributing to delayed healing [53].
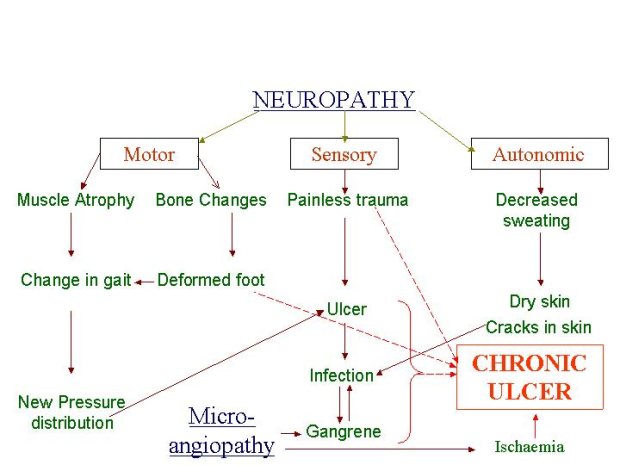
In addition to the generic changes observed in chronic wounds, there are certain disease-specific pathologies that contribute to their non-healing nature (Table 4). For example, in patients with diabetes the chronic nature of foot ulcers is multifactorial (Figure 2): neuropathy and peripheral neuritis causing trophic changes; microangiopathy leading to atheroma of the small arteries resulting in ischaemia, gangrene and infection; and excess sugar lowering resistance to infection. Similarly, in patients with venous ulcers the persistence of ulcers may be due to continued venous hypertension resulting in peripheral oedema. These systemic factors also need to be addressed before healing can be initiated and achieved in such wounds. Some common clinical features and recognised complications of chronic wounds are tabulated in Table 5.
| NEUROPATHY | Diabetes mellitus, Spinal injuries, Cerebral Palsy, Hansen's disease (Leprosy) |
| ISCHEMIA | Atherosclerosis, calcification, microangiopathy (diabetes mellitus), any form of peripheral vascular disease |
| PERIPHERAL OEDEMA | Venous hypertension (deep venous thrombosis, varicose veins), systemic causes (renal or cardiac failure), lymphoedema, decreased albumin, elephantiasis (common in tropical countries) |
| PRESSURE | Poor mobility, spinal cord injuries, dementia, diabetes mellitus, extremes of age, terminal illness |
| OTHER CAUSES | Connective tissue disorders leading to vasculitis, arterio-venous malformations, drugs such as corticosteroids and hydroxyurea, systemic diseases, malignancy, osteomyelitis, smoking, inherited neutrophil disorders, poor nutritional status |
| Clinical features | Complications |
|
|
Wound healing is impaired with advancing age and chronic wounds such as diabetic ulcers, arterial ulcers and CVLUs are more prevalent in older people. In addition to other co-morbidities, the effects of drugs such as non-steroidal anti-inflammatory drugs and steroids, poor mobility (causing pressure ulcers), various alterations in the cellular and molecular characteristics of aged skin can impede the healing process.
With ageing there is a reduced turnover of keratinocytes in the epidermis [54] and a reduction in cell population within the dermis [55]. Microscopic (histological) examination reveals that dermal thickness is reduced after the seventh decade [56]. The rate of epithelialisation declines with age [57] and the in vitro proliferation of keratinocytes grown from aged human donors are reduced in comparison to those isolated from newborn donors [58]. The dermo-epidermal junction also flattens, reducing the proliferative capacity of the epidermal cells and leading to an atrophic appearance. Due to this reduced cellular activity in both the epidermis and the dermis, as well as altered ECM production (see below), older people's skin becomes thinner and less dense, and therefore susceptible to damage even after trivial traumas.
The percentage of senescent fibroblasts in the skin increases with ageing [59]. Apart from their inability to proliferate, senescent fibroblasts show decreased motility [60], an increased latent time [61] and reduced responsiveness to stimulatory growth factors [62]. They also accumulate more fibronectin in the ECM environment due to an increase in the rate of synthesis [63] and an alteration in the physical nature of the fibronectin which causes a decrease in its cell adhesive properties [64], [65]. Ashcroft et al observed that after wounding the formation of granulation tissue was delayed in aged compared to young and middle-aged mice [66]. The impaired formation of granulation tissue may be related to a decrease in fibroblast numbers and collagen density [67].
The ability of fibroblasts to produce ECM proteins and their molecular composition is impaired in older people. With ageing there is a decrease in collagen fibre bundle density with the bundles appearing straighter [62], a reduction and disorganisation of type I collagen [68], an increase in type III collagen [69], a decrease in the HA content [70] and a reduction in the gene expression of elastin [68]. All these factors result in an altered ECM composition that leads to defective proliferative and remodelling phases, impairing wound closure.
There is an up-regulation of MMP-2 in normal aged skin, and MMP-2 and MMP-9 in acute wounds in aged skin in comparison with young adults [71]. This alteration in the cytokine profile is similar to that seen in chronic wounds in younger patients. TIMP-1 and TIMP-2 mRNA levels are also significantly reduced in normal aged skin and after acute wounding in aged skin [72]. These factors may combine to predispose the remodelled wound to recurrent breakdown. In animal models, a delayed inflammatory response has been observed after acute wounding in middle-aged and aged compared to young mice [66]. Similarly, in aged mice a decline in wound macrophage phagocytic function and a delay in T-lymphocyte response in the later stages of healing has been observed [73]. Senescence is also associated with the over-expression of IL-1a in endothelial cells, which inhibits angiogenesis [74] and reduces the expression of IL-6 [75].
In addition to co-morbidities, the clinical impairment of wound healing in the aged may be related to delayed cellular proliferation, changes in ECM production and composition, and an altered cytokine and inflammatory response. These factors, on their own or in combination, contribute to the increased occurrence and longevity of wounds in older people.
It is clear that the environment and healing patterns of acute and chronic wounds, including those in older people, are dissimilar. Because of these differences complete wound closure may not be a realistic outcome in many chronic wounds and wounds in older people. For this reason the use of meaningful alternative endpoints in the assessment of chronic, recalcitrant wounds, such as a reduction in exudate, better pain control or an improvement in overall quality of life, are currently the subject of intense debate.
1. Callam MJ, Ruckley CV, Harper DR, Dale JJ. Chronic ulceration of the leg: extent of the problem and provision of care. Br Med J (Clin Res Ed) 1985; 290(6485): 1855-56.
2. Nelzén 0, Bergqvist D, Lindhagen A, Hallbook T. Chronic leg ulcers: an underestimated problem in primary health care among elderly patients. J Epidemiol Community Health 1991; 45: 184-87.
3. Nelzén O, Bergqvist D, Lindhagen A. The prevalence of chronic lower-limb ulceration has been underestimated: results of a validated population questionnaire. Br J Surg 1996; 83(2): 255-58.
4. Callam MJ, Harper DR, Dale JJ, Ruckley CV. Chronic ulcer of the leg: clinical history. Br Med J (Clin Res Ed) 1987; 294(6584): 1389-91.
5. Clark RAF. Wound repair: overview and general considerations. In: The Molecular and Cellular Biology of Wound Repair. Clark RAF, editor. London: Plenum Press, 1996; 3-50.
6. Clark RA, Lanigan JM, DellaPelle P, Manseau E, Dvorak HF, Colvin RB. Fibronectin and fibrin provide a provisional matrix for epidermal cell migration during wound reepithelialization. J Invest Dermatol 1982; 79(5): 264-69.
7. Grinnell F, Billingham RE, Burgess L. Distribution of fibronectin during wound healing in vivo. J Invest Dermatol 1981; 76(3): 181-89.
8. Leibovich SJ, Ross R. The role of the macrophage in wound repair. A study with hydrocortisone and antimacrophage serum. Am J Pathol 1975; 78(1): 71-100.
9. Slavin J. The role of cytokines in wound healing. J Pathol 1996; 178(1): 5-10.
10. Grinnell F. Fibroblasts, myofibroblasts, and wound contraction. J Cell Biol 1994; 124(4): 401-04.
11. Tonnesen MG, Feng X, Clark RA. Angiogenesis in wound healing. J Investig Dermatol Symp Proc 2000; 5(1): 40-06.
12. Lauer G, Sollberg S, Cole M, Flamme I, Stürzebecher J, Mann K, Krieg T, Eming SA. Expression and proteolysis of vascular endothelial growth factor is increased in chronic wounds. J Invest Dermatol 2000; 115(1): 12-18.
13. Kurkinen M, Vaheri A, Roberts PJ, Stenman S. Sequential appearance of fibronectin and collagen in experimental granulation tissue. Lab Invest 1980; 43(1): 47-51.
14. Clark RA. Wound repair. Curr Opin Cell Biol 1989; 1(5): 1000-08.
15. Clark RAF. Mechanisms of cutaneous wound repair. Volume 1. In: Dermatology in General Medicine. Fitzpatrick TB, Eisen AZ, Wolff K et al, editors. New York: McGraw-Hill, 1993; 473-86.
16. Winter GD. Formation of the scab and the rate of epithelization of superficial wounds in the skin of the young domestic pig. Nature 1962; 193: 293-94.
17. Clark RA, Nielsen LD, Welch MP, McPherson JM. Collagen matrices attenuate the collagen-synthetic response of cultured fibroblasts to TGF-beta. J Cell Sci 1995; 108((Pt 3)): 1251-61.
18. Welch MP, Odland GF, Clark RA. Temporal relationships of F-actin bundle formation, collagen and fibronectin matrix assembly, and fibronectin receptor expression to wound contraction. J Cell Biol 1990; 110(1): 133-45.
19. Levenson SM, Geever EF, Crowley LV, Oates JF, Berard CW, Rosen H. The healing of rat skin wounds. Ann Surg 1965; 161: 293-308.
20. Desmoulière A, Redard M, Darby I, Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol 1995; 146(1): 56-66.
21. Jürgensmeier JM, Schmitt CP, Viesel E, Höfler P, Bauer G. Transforming growth factor beta-treated normal fibroblasts eliminate transformed fibroblasts by induction of apoptosis. Cancer Res 1994; 54(2): 393-98.
22. Lazarus GS, Cooper DM, Knighton DR, Margolis DJ, Pecoraro RE, Rodeheaver G, Robson MC. Definitions and guidelines for assessment of wounds and evaluation of healing. Arch Dermatol 1994; 130(4): 489-93.
23. Yager DR, Zhang LY, Liang HX, Diegelmann RF, Cohen IK. Wound fluids from human pressure ulcers contain elevated matrix metalloproteinase levels and activity compared to surgical wound fluids. J Invest Dermatol 1996; 107(5): 743-48.
24. Schultz GS, Mast BA. Molecular analysis of the environment of healing and chronic wounds: cytokines, proteases and growth factors. Wounds 1998; 10((Suppl F)): 1F-9F.
25. Cook H, Davies KJ, Harding KG, Thomas DW. Defective extracellular matrix reorganization by chronic wound fibroblasts is associated with alterations in TIMP-1, TIMP-2, and MMP-2 activity. J Invest Dermatol 2000; 115(2): 225-33.
26. Grinnell F, Zhu M. Fibronectin degradation in chronic wounds depends on the relative levels of elastase, alpha1-proteinase inhibitor, and alpha2-macroglobulin. J Invest Dermatol 1996; 106(2): 335-41.
27. Wlaschek M, Peus D, Achterberg V, Meyer-Ingold W, Scharffetter-Kochanek K. Protease inhibitors protect growth factor activity in chronic wounds. Br J Dermatol 1997; 137(4): 646.
28. Harris IR, Yee KC, Walters CE, Cunliffe WJ, Kearney JN, Wood EJ, Ingham E. Cytokine and protease levels in healing and non-healing chronic venous leg ulcers. Exp Dermatol 1995; 4(6): 342-49.
29. Trengove NJ, Bielefeldt-Ohmann H, Stacey MC. Mitogenic activity and cytokine levels in non-healing and healing chronic leg ulcers. Wound Repair Regen 2000; 8(1): 13-25.
30. Mendez MV, Raffetto JD, Phillips T, Menzoian JO, Park HY. The proliferative capacity of neonatal skin fibroblasts is reduced after exposure to venous ulcer wound fluid: A potential mechanism for senescence in venous ulcers. J Vasc Surg 1999; 30(4): 734-43.
31. Whiston RJ, Hallett MB, Davies EV, Harding KG, Lane IF. Inappropriate neutrophil activation in venous disease. Br J Surg 1994; 81(5): 695-98.
32. Moore K, Ruge F, Harding KG. T lymphocytes and the lack of activated macrophages in wound margin biopsies from chronic leg ulcers. Br J Dermatol 1997; 137(2): 188-94.
33. Loots MA, Lamme EN, Zeegelaar J, Mekkes JR, Bos JD, Middelkoop E. Differences in cellular infiltrate and extracellular matrix of chronic diabetic and venous ulcers versus acute wounds. J Invest Dermatol 1998; 111(5): 850-57.
34. Stanley AC, Park HY, Phillips TJ, Russakovsky V, Menzoian JO. Reduced growth of dermal fibroblasts from chronic venous ulcers can be stimulated with growth factors. J Vasc Surg 1997; 26(6): 994-99; discussion 999-1001.
35. Agren MS, Steenfos HH, Dabelsteen S, Hansen JB, Dabelsteen E. Proliferation and mitogenic response to PDGF-BB of fibroblasts isolated from chronic venous leg ulcers is ulcer-age dependent. J Invest Dermatol 1999; 112(4): 463-69.
36. Mendez MV, Stanley A, Phillips T, Murphy M, Menzoian JO, Park HY. Fibroblasts cultured from distal lower extremities in patients with venous reflux display cellular characteristics of senescence. J Vasc Surg 1998; 28(6): 1040-50.
37. Vande Berg JS, Rudolph R, Hollan C, Haywood-Reid PL. Fibroblast senescence in pressure ulcers. Wound Repair Regen 1998; 6(1): 38-49.
38. Adair HM. Epidermal repair in chronic venous ulcers. Br J Surg 1977; 64(11): 800-04.
39. Andriessen MP, van Bergen BH, Spruijt KI, Go IH, Schalkwijk J, van de Kerkhof PC. Epidermal proliferation is not impaired in chronic venous ulcers. Acta Derm Venereol 1995; 75(6): 459-62.
40. Agren MS, Eaglstein WH, Ferguson MW, et al. Causes and effects of chronic inflammation in venous leg ulcers. Acta Derm Venereol Suppl (Stockh) 2000; 210: 3-17.
41. Bucalo B, Eaglstein WH, Falanga V. Inhibition of cell proliferation by chronic wound fluid. Wound Repair Regen 1993; 1(3): 181-86.
42. Raffetto JD, Mendez MV, Marien BJ, Byers HR, Phillips TJ, Park HY, Menzoian JO. Changes in cellular motility and cytoskeletal actin in fibroblasts from patients with chronic venous insufficiency and in neonatal fibroblasts in the presence of chronic wound fluid. J Vasc Surg 2001; 33(6): 1233-41.
43. Herrick SE, Sloan P, McGurk M, Freak L, McCollum CN, Ferguson MW. Sequential changes in histologic pattern and extracellular matrix deposition during the healing of chronic venous ulcers. Am J Pathol 1992; 141(5): 1085-95.
44. Herrick SE, Ireland GW, Simon D, McCollum CN, Ferguson MW. Venous ulcer fibroblasts compared with normal fibroblasts show differences in collagen but not fibronectin production under both normal and hypoxic conditions. J Invest Dermatol 1996; 106(1): 187-93.
45. Hasan A, Murata H, Falabella A, Ochoa S, Zhou L, Badiavas E, Falanga V. Dermal fibroblasts from venous ulcers are unresponsive to the action of transforming growth factor-beta 1. J Dermatol Sci 1997; 16(1): 59-66.
46. Salim AS. The role of oxygen-derived free radicals in the management of venous (varicose) ulceration: a new approach. World J Surg 1991; 15(2): 264-69.
47. Abd-El-Aleem SA, Ferguson MW, Appleton I, Kairsingh S, Jude EB, Jones K, McCollum CN, Ireland GW. Expression of nitric oxide synthase isoforms and arginase in normal human skin and chronic venous leg ulcers. J Pathol 2000; 191(4): 434-42.
48. Howlader MH, Smith PD. Increased plasma total nitric oxide among patients with severe chronic venous disease. Int Angiol 2002; 21(2): 180-86.
49. Jude EB, Tentolouris N, Appleton I, Anderson S, Boulton AJ. Role of neuropathy and plasma nitric oxide in recurrent neuropathic and neuroischemic diabetic foot ulcers. Wound Repair Regen 2001; 9(5): 353-59.
50. Falabella AF, Carson P, Eaglstein WH, Falanga V. The safety and efficacy of a proteolytic ointment in the treatment of chronic ulcers of the lower extremity. J Am Acad Dermatol 1998; 39(5 Pt 1): 737-40.
51. Baharestani M. The clinical relevance of debridement. In: The clinical relevance of debridement. Baharestani M, Gottrup F, Holstein P, Vanscheidt W, editors. Heidelberg: Springer-Verlag, 1999; 1-13.
52. Davey ME, O'toole GA. Microbial biofilms: from ecology to molecular genetics. Microbiol Mol Biol Rev 2000; 64(4): 847-67.
53. Ladwig GP, Robson MC, Liu R, Kuhn MA, Muir DF, Schultz GS. Ratios of activated matrix metalloproteinase-9 to tissue inhibitor of matrix metalloproteinase-1 in wound fluids are inversely correlated with healing of pressure ulcers. Wound Repair Regen 2002; 10(1): 26-37.
54. Grove GL, Kligman AM. Age-associated changes in human epidermal cell renewal. J Gerontol 1983; 38(2): 137-42.
55. Kurban RS, Bhawan J. Histologic changes in skin associated with aging. J Dermatol Surg Oncol 1990; 16(10): 908-14.
56. Shuster S, Black MM, McVitie E. The influence of age and sex on skin thickness, skin collagen and density. Br J Dermatol 1975; 93(6): 639-43.
57. Holt DR, Kirk SJ, Regan MC, Hurson M, Lindblad WJ, Barbul A. Effect of age on wound healing in healthy human beings. Surgery 1992; 112(2): 293-97; discussion 297-8.
58. Gilchrest BA. In vitro assessment of keratinocyte aging. J Invest Dermatol 1983; 81(1 Suppl): 184s-9s.
59. Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 1995; 92(20): 9363-67.
60. Kondo H, Yonezawa Y. Changes in the migratory ability of human lung and skin fibroblasts during in vitro aging and in vivo cellular senescence. Mech Ageing Dev 1992; 63(3): 223-33.
61. Muggleton-Harris AL, Reisert PS, Burghoff RL. In vitro characterization of response to stimulus (wounding) with regard to ageing in human skin fibroblasts. Mech Ageing Dev 1982; 19(1): 37-43.
62. Ashcroft GS, Horan MA, Ferguson MW. The effects of ageing on cutaneous wound healing in mammals. J Anat 1995; 187((Pt 1)): 1-26.
63. Shevitz J, Jenkins CS, Hatcher VB. Fibronectin synthesis and degradation in human fibroblasts with aging. Mech Ageing Dev 1986; 35(3): 221-32.
64. Chandrasekhar S, Sorrentino JA, Millis AJ. Interaction of fibronectin with collagen: age-specific defect in the biological activity of human fibroblast fibronectin. Proc Natl Acad Sci U S A 1983; 80(15): 4747-51.
65. Porter MB, Pereira-Smith OM, Smith JR. Novel monoclonal antibodies identify antigenic determinants unique to cellular senescence. J Cell Physiol 1990; 142(2): 425-33.
66. Ashcroft GS, Horan MA, Ferguson MW. Aging is associated with reduced deposition of specific extracellular matrix components, an upregulation of angiogenesis, and an altered inflammatory response in a murine incisional wound healing model. J Invest Dermatol 1997; 108(4): 430-37.
67. Kligman AM, Lavker RM. Cutaneous aging: the differences between intrinsic aging and photaging. J Cutan Aging & Cosm Dermatol 1998; 1(1): 5-12.
68. Uitto J, Fazio MJ, Olsen DR. Molecular mechanisms of cutaneous aging. Age-associated connective tissue alterations in the dermis. J Am Acad Dermatol 1989; 21(3 Pt 2): 614-22.
69. Lovell CR, Smolenski KA, Duance VC, Light ND, Young S, Dyson M. Type I and III collagen content and fibre distribution in normal human skin during ageing. Br J Dermatol 1987; 117(4): 419-28.
70. Fleischmajer R, Perlish JS, Bashey RI. Human dermal glycosaminoglycans and aging. Biochim Biophys Acta 1972; 279(2): 265-75.
71. Ashcroft GS, Horan MA, Herrick SE, Tarnuzzer RW, Schultz GS, Ferguson MW. Age-related differences in the temporal and spatial regulation of matrix metalloproteinases (MMPs) in normal skin and acute cutaneous wounds of healthy humans. Cell Tissue Res 1997; 290(3): 581-91.
72. Ashcroft GS, Herrick SE, Tarnuzzer RW, Horan MA, Schultz GS, Ferguson MW. Human ageing impairs injury-induced in vivo expression of tissue inhibitor of matrix metalloproteinases (TIMP)-1 and -2 proteins and mRNA. J Pathol 1997; 183(2): 169-76.
73. Swift ME, Burns AL, Gray KL, DiPietro LA. Age-related alterations in the inflammatory response to dermal injury. J Invest Dermatol 2001; 117(5): 1027-35.
74. Maier JA, Voulalas P, Roeder D, Maciag T. Extension of the life-span of human endothelial cells by an interleukin-1 alpha antisense oligomer. Science 1990; 249(4976): 1570-74.
75. Goodman L, Stein GH. Basal and induced amounts of interleukin-6 mRNA decline progressively with age in human fibroblasts. J Biol Chem 1994; 269(30): 19250-55.